

본 게시글은 학부 '생물정보학' 강의를 수강한 내용을 토대로 이해한 바를 정리하였습니다.
흐름 잘타야 한다. 여기를 잘 이해하느냐가 성적을 좌우할 것이다.
- Neverthe1ess

우리는 수능의 민족. 중학교 고등학교를 거쳐 수능을 치르고 대학교에 입학했다. 우리는 공부를 더 잘하고 싶다. 우리는 각자 개개인이 공부하고 싶은 과목이, 잘하는 과목이 있었을 것이다. 하지만 우리는 수능을 치르기 위해서는 하기 싫은 과목도 공부해야 했다. 그랬기에, 우리의 성적은 전체적으로 하향평준화가 되었을지도 모른다. 이와 달리 대학교에서는 우리가 듣고 싶은 과목을 골라 들을 수 있다.(물론 빠른 클릭과 타이밍을 재는 능력은 필수.)
공부를 오래한 사람과 그렇지 않은 사람. 한 분야에 오래 종사한 사람과 그렇지 않은 사람. 여러 차이가 있겠지만 앞의 두 부류의 가장 명확한 특징적인 차이는 무엇일까?
익숙함
익숙하다는 것은 어떻게 설명해야 할까? 머릿속에 이미 기본 탑재가 되어있느냐 안 되어있느냐의 차이가 아닐까?
예를 들어, 우리가 영어를 공부할 때도 " I'm sorry " 또는 " Thank you " 라는 문구를 말하거나 읽을 때, 우리는 이를 지금 따로 해석을 하고 있는가?
아닐 것이다. 뇌를 거치지 않는다. 어떻게 보면 우리의 한국말과 비슷한 포지션을 가지게 된 문구일 것이다. 하지만 문법이 점점 어려워지고 문장이 길어지면 우리는 그때부터 대화부터 읽기까지 점점 버벅거리기 시작한다. 그 긴 문장은 내 머릿속에 탑재 되어있지 않아 익숙하지 않다는 것이다.
물론 신이 내려준 능력으로 인한 차이도 있을 것이다. 어려운 개념을 한 번에 이해한 사람이 있는 반면 여러 번의 설명을 듣고, 반복을 해야만 비로소 이해가 되는 사람도 있을 것이다. 이러한 상태를 본인 스스로 파악하는 것을 '메타인지'라고 하지 않은가?
결국.. 우리에게 재미없는 과목은 안 들을 것이다. 안듣게 되면 계속 못하게 될 것이다. 안타깝다. 재미없는 과목에 시간을 더 투자해서 잘해져야 하는 시국에 나는 더 안하고 있다....
심지어 이 생물정보학은 학부의 모든 생물학도에게 생소할 것이다. 이 과목에 흥미를 느낀다면 좋겠지만, 설령 재미없더라도 이 시대에 필요한 과목인만큼 잘해져야 한다. 그래서 마인드세팅이 필요하다. 목적의식을 만들어야 할 것이다.
빅데이터가 무엇인지 와닿는다면 내가 무엇을 해야할 지 느낄 수 있지 않을까? 나도 한 번의 수업으로 바이오 빅데이터를 이해하지 못했다. 지금도 그렇다. 하지만 어떤 범주에 속해있고 어떤 것들이 있는지 구성 요소들을 알면 목적의식이 생길 것이다.
반복 숙달을 통해 익숙해져야 한다.
핵 안에 DNA가 있고 DNA 안에 유전 정보가 있다. 그 유전 정보를 통해서 mRNA를 만들고 이를 통해 단백질을 만들어서 이들이 흩어져서 일을 하는... 식물이나 동물이나 근본은 비슷하다. 그래서 식물, 동물... 생물정보학은 크게 다른게 없다. 우리는 더 안에 있는 깊이 있는 데이터를 다룰 것이다. 물론 다른 생리적인 특징을 알고 있다면 도움은 될 것이다. 분석을 했을 때 차이나는 부분이 크게 없다는 것이다.
바이오 빅데이터의 범주
염색체 수가 몇 개인지 셀 수 있고, 배수성이있는지 없는지 볼 수 있고, Kryotyping 분야가 할 수 있는게 굉장히 많다. 아래의 그림과 같은 염색체는 아주 찰나의 순간에 지나가는 상태이다. 대부분 풀어헤쳐진 상태로 존재한다. 유전자는 전체 DNA 주우에 인간의 경우 2% 밖에 되지 않는다. 그 2% 중에 RNA polymerase가 와서 유전자를 transciption을 할 것이다. RNA polymerase는 크기가 매우 크다. 그 친구가 유전자에 앉으려면, Binding을 하려면 그 자리에는 히스톤이 떡하니 지키고 있다. 히스톤이 지키고 있는 한, 히스톤으로 DNA가 감겨있으면 RNA polymerase는 binding 할 수 없다.
그래서 풀려야 한다. 이를 연구해서 우리는 어디가 methylation이 되면 안풀리고, 어디가 acylation이 되면 잘 풀리는지를 안다. 이를 과거에는 실험을 통해 밝혀야 했지만 이제는 실험을 거치지 않아도 알 수 있게 되는 부분이 많아지고 있다. 그래서 연구 속도는 더욱 빨라지고 있다.
Genomics
- 유전체 해독 : Gene annotation. DNA의 정보가 없으면 만들어내는 일. ex) Human Genome Project
- 유전체 조립 : De novo Assembly 또는 Genome Assembly. 단순히 DNA 서열을 알아내는 것.
유전체 해독은 조립하고, 어떠한 유전자의 영역 중 exon은 어디에 있는지, intron은 어디에 있는지 등 까지 알아내는 것. 뉘앙스의 차이가 있음을 알자.
유전체 조립은 단순 서열을 연결시키는 일이다. 해독은 Gene annotation까지 진행해야 해독이라는 의미가 있다.
인간 Genome 30억개를 본다고 하면, 그냥 단순히 A, T, G, C 만 있을 것이다. 하지만 그냥 이를 봐서 뭐하는가. 이 Gene에 대한 정보가 하나도 없지 않은가. 그냥 DNA를 처음부터 끝까지 쭉 읽고 싶으면 DNA sequencing을 하면 된다. 앞서 설명한 PacBio, Nanopore를 사용해서 말이다.
정확한 read가 필요하면 글룸, short read 등도 같이 써야할 것이다. 그래서 요즘은 그 2가지를 같이 쓰는 Hybrid Assembly방식으로 진행한다. DNA sequencing을 한다는 것은 유전체 정보를 만들겠다는 뜻이다. 이 때는 Long-read가 반드시 필요하다. 이제 기술이 많이 좋아져서 가격도 그만큼 내려가 동시에 여러가지를 작업할 수 있게 되었다.
그래서 (PAN) genome이 가능해졌다.
- (PAN) Genome : 같은 속, 종 내의 여러 DNA를 동시에 조립하는 것. 완벽하게.
예를 들어, 안경 쓴 집단과 안경을 쓰지 않은 집단을 나누어 이들을 human genome project를 진행해서 30억 개를 맞춰보는 것이다. 과연 왜 안경을 쓰게 되었는지 전체 DNA를 비교할 수 있는 것이다. 하나도 빈틈없이 비교한다.
같은 속 내의 다른 종들끼리 비교하는 것을 종간비교라고 한다. 그리고 같은 종 내의 다른 대표 선수들 간의 비교는 종내비교 라고 한다. 그래서 이런 모든 gonome vs genome 비교를 PAN Genome 이라고 하는 것이다.
그 결과 우리는 좀 더 디테일하고 확실하게 비교할 수 있다. 그래서 이를 진화 분석에 사용이 가능해진다.
- DNA-reseq : SNP만 찾기 위함. 전체를 조립하지 않는다.
물론 DNA 전체를 비교하는 만큼 비용이 많이 든다. 그래서 비용절감 차원에서 DNA-reseq을 사용한다. Resequencing을 한다.
- DNA-seq : 유전체, 전체를 조립 하겠다. Whole genome seqeuncing. 전체 새로운 DNA를 조립해서 정보를 만든다.
DNA-reseq의 경우 SNP만을 찾으려면 기준이 있어야 한다. 그 기준을 표준 유전체 라고 한다. 선생님 한 명과 여러 학생들이 있다면 선생님이 표준 유전체라고 보면 되겠다. 선생님, 즉 그 표준 유전체는 '완벽한' 하나여야 한다.
- GBS : Genotyping by Sequencing. 비용 절감 차원.
Genotyping은 SNP를 찾는 걸 일컫는다. 유전형을 찾아내는 것. GBS의 경우 제한 효소로 DNA를 다 자른다. 우리는 모두 Human 이니까 제한 효소의 절단 부위는 다 비슷할 것이다. 서로 잘린 부위만 읽는다. 그 잘린 부위만 alignment를 진행한다.
여기 까지 정리
- PAN Genome : 전체 DNA를 찾기 위한 목적
- DNA-reseq & GBS : 유전 변이만 찾기 위한 목적
Bisul-seq(바이설파이트 시퀀싱, DIPseq(딥시퀀싱))
DNA가 왜 풀리는 지를 알기 위해서는 Methylation을 이해해야 한다. 이를 보기 위해 진행하는 것이 Bisul-seq 또는 DIPseq.
Histone modification
특정 아미노산의 항체를 쓰게 되면 그 부위가 어딘지 어디서부터 어디인지, 그 DNA를 읽는다. 이 Histone은 동물, 식물들에 모두 흔하게, 공통적으로 쓸 수 있다. 그 DNA를 읽어서 어떤 Histone에 영향을 주었는지를 알 수 있는 방법이다.
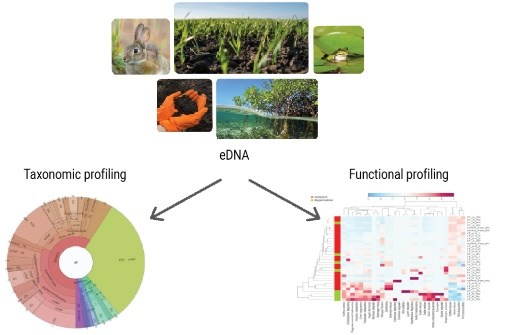
Metagenome
미생물 전부. 흙에 있는 미생물, 물에 있는 미생물 또는 장내 미생물. 식물 안에 있는 미생물도. 이러한 미생물은 우리가 키울 수 없다. 키울 수는 없지만, 그 안에 존재한다면 그 DNA를 추출해서 sequencing을 할 수 있는데 앞서 설명한 seq과는 차이가 있다. 앞서 언급한 DNA seq은 eukaryote, 다시 말해 핵이 존재하고 염색체가 존재하는 반면 이 미생물들은 DNA가 동그랗다. 그리고 DNA 종류가 매우 많다. 한 종이 아니라는 것. 따라서 군집 분석 밖에 할 수 없다. 길게 읽게 되면 정보도 만들어낼 수 있다. 미생물에 대한 정보를 만드는 것. 예를 들어 군집분석이라 하면, 어떠한 커뮤니티가 어떠한 토양에는 어떤 종이 우점하고 있고, 어떠한 물 속에는 어떤 미생물이 우점하고 있는지 나의 장 속에는 어떤 미생물이 우점하고 있는 지 등의 군집 분석을 한다. 내가 과민성 대장 증후군을 앓고 있다면 분명 정상인들과는 다른 장 미생물을 가지고 있을 것이다. 이들을 치환해줌으로서 치료를 할 수 있는 방법론을 제시할 수도 있는 것이다.
Transriptomics
이 sequencing은 비교적 쉽다. RNA seq과 Non coding RNA seq 만 있기 때문이다. 최근들어 single cell RNA seq(scRNA seq)이 생겼다. 하지만 원리는 똑같다.
- RNA-seq : 조직, 기관에 해당하는 세포를 다 싸잡아서 RNA 정량
- scRNA-seq : 암세포가 있다면 이를 다 분리해서 세포 하나하나 다 들여다 보는 것. 요즘 트렌드.
scRNA-seq의 경우 이제 Bulk-RNA seq이라 해서 그냥 싸잡아서 분석한다.
- Quant-seq : 유전자의 발현량, 정량적 분석. 유일하게 Single-end read로 생산. 표준유전체가 필요없다.
Chip-seq
Chromatic IP. Transciption factor, 전사 조절 인자. RNA polymerase가 오기 전에 먼저 기다리고 있는 물질이다. 이 전사 조절 인자에 따라서 뒤에 발현하는 유전자가 조절된다. 즉, 그 transcription factor가 어디에 binding 하는지 중요할 것이다. 어디에 binding하는지 알면 그 타겟 유전자를 찾을 수 있다.
Transciption factor를 왜 연구하느냐. Transcription factor는 유전자를 발현시키기 위해 한 자리에만 binding 하는 것으로 알고 있었지만 그게 아니었음이 밝혀졌다. 전체 DNA 상의 무수히 많은 곳에 앉아있던 것. 어떤 때에 거기에 앉고, 어떤 때에는 다른 곳에 앉는다는 것이다. Transcription factor를 연구하면 좋은 이유가 유전자 하나늘 발현시켰을 뿐인데 그 하나로 인해서 다양한 유전자가 동시에 발현되는 효과가 있다. 키의 경우 다인자유전인 것으로 유명한데, 이에 관여하는 유전자가 15곳이 있다고 하면 15개를 하나하나 씩 넣는 것보다 15개를 한 번에 조절하는 전사 조절 인자 하나를 넣는게 훨씬 유용할 것이다. 그래서 학계에서는 Transcription factor 연구에 열정적인 것.
내가 관심있는 transcription factor가 어디에 binding 하는지를 알면 그 다음 타겟이 무엇인지 알 수 있을 것이다. 그리고 어떤 유전자를 컨트롤하는지도 알 수 있을 것이다. 이를 위해서 Chip-seq을 진행한다. 이것도 마찬가지로 레퍼런스, 표준 유전체가 없으면 수행할 수 없다.
ncRNA-seq(Non coding RNA-seq)
non coding RNA는 사이즈 별로 다르다. siRNA, miRNA 같은 경우는 22 bp ~ 23 bp, 길어야 24 bp 정도 된다. 이들을 모아 sequencing을 하는 방법도 있지만, 요즘은 이러한 short non coding 보다 long non coding RNA에 더 관심이 많다.
이러한 long non coding RNA를 줄여서 lncRNA라고 한다. 하지만 lncRNA를 그냥 읽을 수는 없기 때문에 Total RNA-seq을 이용해야 한다.
Total RNA-seq은 non coding RNA와 Bulk RNA를 모두 포함한다는 말. 즉, mRNA 뿐 아니라 non coding RNA 다 나온다. Quant-seq을 넘어 bulk RNA-seq은 유전자 발현량 뿐만 아니라 유전자의 서열도 알 수 있다.
Bulk RNA-seq은 표준 유전체 레퍼런스가 없어도 할 수 있다. 유전자 정보를 scRNA-seq를 통해서 알 수 있기 때문이다. 이미 우리가 관심있는 조직을 알고 있을 것. 반면에 Quant-seq은 유전자 서열에 대한 정보는 알 수 없다. 그래서 Quant-seq의 경우 무조건 표준 유전체가 있어야 한다. 유전체 조립을 할 수 있는 여건이 안된다면 bulk RNA-seq을 진행하면 된다.
Proteomics
Protein-seq
LC-MS 사용 : 이미 데이터베이스가 있는 peptide 정보를 이용해서 원하는 peptide 정보가 나오면 이를 맞춘다.
Nanopore 사용 : 단백질을 Denaturation을 시켜서 nanopore를 통과하면 전류의 차이를 읽어낸다.
Metabolomics
대사물질을 분석한다. 1차 대사물질, 2차 대사물질 등.
- LC-MS
- GS-MS
- NMR
아직 sequencing은 없다. 구조적으로 Sequencing과 어울리지 않는다. 정확히는 있을 수가 없다. Sequencing은 서열, 순서가 있어야 하는데 이 물질들은 단지 화학 구조. 따라서 Sequencing 기술이 있을 수가 없다. 심지어 Lipid가 포함된다. 그래서 연구하기가 상당히 까다롭다.
Phenomics
사진 데이터(이미지 데이터)
- 3D point cloud : 점으로 되어있는 데이터(Point Cloud). 이를 3D LiDAR 장비로 촬영
- RGB : 카메라. 시중 휴대폰 카메라도 상당한 Phenotype data를 얻을 수 있다. 이를 이용해 3D로 만들 수 있다.
- RS Images : RS(Remote control System, Sensing). Spectral image. 긴, 짧은 파장을 이용해서 세포나 식물, 동물을 분석할 수 있는 이미지 데이터
여기까지를 잘 이해하고 있으면 어떤 데이터를 어떨 때 사용해야 되는지 그리고 그 데이터는 어떻게 생산을 해야 되는지, 무엇을 이용해서 생산해야 하는지 등을 판단하는데 상당한 도움이 될 것이다.
'BIOLOGY > Bioinformatics' 카테고리의 다른 글
| 데이터 만들기 a.k.a 데이터 생산 (3) | 2024.10.19 |
|---|---|
| Sequencing에 점수를, Phred score (0) | 2024.10.18 |
| 생명과학의 미래, NGS(차세대 염기서열 분석) (2) | 2024.10.15 |
| 바이오 빅데이터의 종류와 활용 (4) | 2024.10.13 |
| 생물학, 그리고 코딩을 곁들인 (1) | 2024.09.06 |
나의 성장 드라마
포스팅이 좋았다면 "좋아요❤️" 또는 "구독👍🏻" 해주세요!



