생명과학의 미래, NGS(차세대 염기서열 분석)
by Ungbae본 게시글은 학부 강의 '생물정보학(Bioinformatics)'를 수강한 내용을 토대로 필자가 이해한 점들을 정리하였습니다.
Sequencing(시퀀싱, 염기서열 분석)

- DNA sequence

- RNA sequence

Sequencing의 역사
1977년 Sanger의 Sanger method 개발
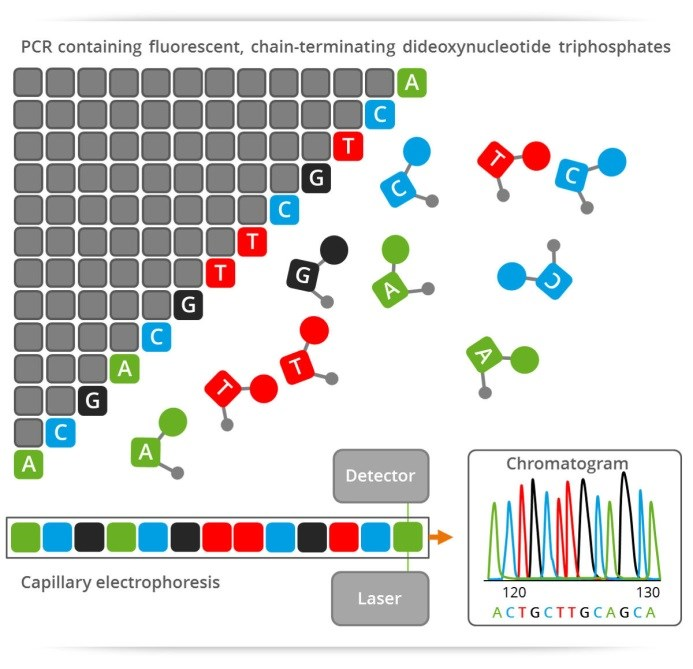
분자생물학이라는 학문이 생기면서 우리는 DNA 서열을 모르면 분자생물학을 제대로 이해할 수 없게 되었다. 그 가장 흔한 Cloning 실험 조차도 우리는 서열을 모르면 진행할 수 없다. 우리가 배우는 Enzyme site. 제한 효소가 자르는 자리를 알기위해서는 Sequencing 기술이 있기에 가능하다. 우리가 아무 생각없이 행해왔던 실험은 모두 sequencing이라는 기술이 있었기에 지금의 모든 학문이 깊어지고 있다고 봐도 무방할 것이다.
하지만 1990년 Human genome project가 시작되면서 기존의 sequencing 기술이 현대의 연구 주제를 모두 커버하기에는 너무 느리다라는 것을 학계가 점점 자각하게 되면서 새로운 방법들을 강구해 나갔다. 30억개나 되는 DNA를 알아내기에는 너무 많은 시간이 걸릴 것이다. 그래서 우리는 다음 세대, Next Generation의 기술이 도입되어야 한다.
그렇다 NGS 기술들의 등장이다.
NGS(Next Generation Sequencing)

동시에 수천만 개의 DNA를 동시에 읽을 수 있는 기술을 세계는 보유하고 있다. 하나의 DNA를 알면 이 DNA는 어떠한 서열인지 매칭이 된다는 것. DNA를 한 번 읽게 되면 데이터는 어마어마하게 쏟아져 나오는데 이 데이터가 누구 것이니지 바로 바로 판단이 되어야 할 것이다. 지도의 위도와 경도처럼 좌표가 필요하다.
다시 말해, DNA를 동시에 읽게 되면 그 수천만의 데이터, 수천만의 좌표가 나오게 된다. 이렇게 NGS는 탄생했다.
NGS 파헤치기 : DNA 편

1번 과정, DNA extraction
DNA를 준비하는 과정. DNA는 상태가 매우 좋아야 한다. 상태가 좋다는 뜻은 깨지지 않고 깨끗하고 불순물이 없음을 뜻한다. 여기서 간과하면 안되는 점은 개체에 있는 세포들은 모두 같은 DNA를 가지고 있다는 것이다. 다시 말해, 우리의 머리카락 DNA와 피부의 DNA가 다르지 않다는 것. 혈액에 있는 DNA도 똑같다. 다르면 안된다. 물론 Somatic Variation(체세포 변이)의 경우 일부 다른 경우가 생길 수 있다. 예를 들면, 피부세포는 자외선을 받다보면 티민 이량체(Dimer)가 생기는데 이것이 그 예라고 볼 수 있다.
엄밀히, 정확하게 말하면 우리는 모두 각각 아주 일부 다른 DNA를 갖고 있다고 봐야한다. 하지만 그 다름이 매우 미약하기 때문에 다 같은 DNA를 가졌다라고 통상적으로 이야기한다.
RNA의 경우 mRNA를 보면 단백질이 필요할 때만 만들어진다. Cell에 대해 specific하다는 것이다. 우리 몸에는 약 200종류의 세포 종류가 존재한다. 즉, 세포마다 자기가 필요한 단백질은 다르다는 것이다. 어떤 tissue를 하느냐가 매우 중요하게 된다. 간암을 연구하려면 간에서 발현하는 유전자를 중심으로 봐야하지 폐에서 발현한 유전자를 써봐야 의미 없을 확률이 더 크다는 것.
따라서, RNA seq과 DNA seq은 구분해야 한다.
2번 과정, Library preparation
DNA는 어디를 해도 상관이 없다. DNA는 결국 가장 깨끗하게 잘 나오는 곳을 골라 추출하면 된다. 다시 말해, 샘플링하기 편한 곳을 찾으면 된다. 구강세포면 구강세포, 혈액이라면 혈액을 해도 상관이 없다. 그리고 DNA를 잘 추출한 다음 일정한 크기로 자른다.
일정한 크기로 자르는 이유 : DNA를 촘촘히 읽을 수 있다.
2번 과정 그림에서 Adapter가 있다. Adapter를 양쪽에 붙여주는 데 이는 텔로미어와 같은 역할을 수행한다. Primer, DNA가 합성을 하려면 Polymerase가 앉을 자리가 필요하다. 증폭을 위해서, 그러한 이유로 adapter가 있어야 온전한 DNA가 읽힌다.
따라서 이 Adapter를 붙인 결과물을 DNA Library 라고 한다.
이 과정은 매우 중요하다. Library, 말그대로 도서관을 만드는 것이다. 모든 DNA가 들어있는 건데, Library를 잘못만들게 되면 편협한 DNA만 나오게 되는 것. 그래서 포괄적인 DNA를 잘 만들려면 DNA를 깨끗하게 추출해야 똑같은 크기로 잘려서 다양한 DNA를 품게 된다.
위의 2번 과정을 보게 되면 Adapter가 붙은 Library DNA 한 가닥가닥 floor cell에 심게 된다. 이는 기계가 진행한다. 하지만 그 작은 DNA에서 형광을 관찰하려면 빛이 너무 약할 것이다. 그래서 이를 증폭시켜야 한다.
3번 과정, DNA Library sequencing
A, G, C, T가 각각 다른 빛을 갖고 있는데, Cluster를 만들어 주어야 한다.
** 매우 중요**

위의 그림에서 Data collection을 보자. 이 부분이 NGS에 있어서 매우 중요하다. 앞에서 설명하였듯이 NGS는 수천만개를 동시에 읽는다고 언급하였다. Data collection 결과에서 각각 다른 색의 빛은 하나의 좌표가 된다. 2번 과정의 floor cell을 수직, 위의 시선에서 보았다고 생각하면 된다. 따라서, data collection에서 관찰되는 빛은 수천만개라는 것이다. 예를 들어 파란색이 번쩍하면 A T G C 중 C라는 것이고 C가 합성되었음을 의미한다. 만약 A가 합성되었다면 빨간색이 검출된다는 것이다. 이를 통해 검출된 것들을 종합하여 DNA를 한 가닥으로 만든다.
원래 DNA는 두 가닥이지 않은가. 1가닥으로 만들어서 한 번 Washing을 진행한 다음 A, T, G, C를 쫙 뿌려준다. 그러면 밑에서부터 차례대로 상보적인 염기가 붙게 될 것이고 DNA replication 과정처럼 Helicase가 벌려주고, priming site에서 polymerase가 붙어 연결하는 등의 과정을 진행할 것이다.
씻고 다시 붙이고, 씻고 다시 붙이고.... 이를 반복해서 100번 하면 DNA 서열 100개를 읽을 수 있다는 것이다. 밑에서부터 위까지. 이 과정을 또 여러 번 진행하게 되면 100 bp의 DNA 정보가 수천만 개가 나올것이다. 그 수천만개는 랜덤하게 이루어졌기 때문에(마구잡이로 잘랐음을 떠올려라) 그 뒤에 어떻게 나열되는지 조립을 해야한다.
4번 과정, Data analysis
Assembled sequence가 없다고 생각하면, 처음부터 DNA가 없었다고 가정하면... 이제 우리가 직소 퍼즐처럼 끼워 맞추는 작업을 시작해야 한다. 끼워 맞추는 것이다.

이 그림을 보면 중간 중간 오버랩 되는 부분을 발견할 수 있다. 이 오버랩 부분을 따라서 맞추다보면 원래의 DNA 서열이 된다. 1번에 하나 밖에 못읽었던 Sanger method와 비교해보자. NGS는 단 한 번에 모두 읽을 수 있다.
Sanger method와 NGS의 차이는 1번에 하나씩 생산하느냐, 동시에 대량으로 생산하느냐로 볼 수 있다.
NGS 파헤치기 : Protein 편

Protein sequencing. DNA sequencing과 RNA sequencing은 방법이 똑같다. 하지만 Protein sequencing은 다르다.
Protein sequencing의 경우 단백질을 먼저 뽑는다. 단백질을 추출하고 단백질도 잘게 자르는데 여기에서도 룰이 있다. 아무거나 enzyme를 써서 자르면 안된다. 정해진 enzyme을 사용해야 한다. 대표적으로 트립신. 트립신은 자르는 아미노산이 정해져 있다. 그곳만 자른다는 것. 내가 알고 있는 단백질이 있다고 하자. 내가 알고 있는 단백질을 트립신으로 자른다. 그리고 위의 그림에도 있듯이 LC-MS 장비를 사용해서 자른 단백질을 넣으면 펩타이드 하나하나의 질량값이 나온다. 그 질량을 다 다하면 단백질의 이름이 나오게 된다. 질량을 구해 저장된 Database이다. 즉, 이미 데이터 베이스가 현재 구축이 되어있고 이 기존의 데이터베이스의 아미노산 서열이 없으면 찾을 수 없다. 다행히 요즘은 거의 다 분석이 되어있다. 그래서 단백질의 펩타이드를 자른 다음 LC-MS를 이용하면 길이별로, 무게별로 분리가 된다. Liquid chromatography이기 때문에 액상을 타고 쭉 분리가 된다.

단백질을 연구하는 사람들은 미지의 단백질 서열은 모른다. 알 수가 없다. 데이터 베이스에 있어야만 알 수 있다는 것이다. 하지만 열을 가해 만들어진 1차 구조의 단백질을 20개 아미노산의 전기적인 힘을 가지고 아미노산을 읽는 기술이 드디어 나왔다!! Nanopore에 집어 넣어 알아내는 방식이다. 그래서 요즘은 아미노산을 그냥 sequencing을 한다. 단백질을 풀어헤쳐 single molecule로 만들고 nanopore, 나노 스케일의 아주 작은 구멍에 집어 넣으면 기기 안의 current 전류가 있어서 아미노산마다 존재하는 고유 전류를 읽는다. 이제 아미노산도 DNA 서열 읽듯이 읽을 수 있게된 것이다.
대체 저걸 어디에 써먹는거지?
결국 의료기술 아니겠는가. 이걸 이용해서 신약 개발 속도도 늘어났다.
mRNA를 sequencing 하면 암세포에만 독특하게 튀어나와 있는 유전자를 찾을 수 있을 것이다. 암에만 튀어나와 있는. 그것이 항원 아닌가. 그러면 이것을 이용해서 역으로 항체를 만든다. T cell에 박아주는 것. 그럼 그 T cell은 항원만 찾아갈 것이고 그 항원만 죽인다. T cell보다 더 강한 NK cell에 또 이를 적용하면 더 효과적일 것이다. 그리고 이것이 실제로 성공했다. CAR-T가 그래서 세포에 면역 항체를 달아주고 그 T cell이 그 항원만 있는 곳에 쫓아가서 먹다 보니 암세포가 사라졌다.
포인트는 세포의 안도 아니고 그 바깥의 튀어나오는 항원을 찾는 기술.
Zhang et al. 2023 on CAR-T cell functionality improvements and its applications in patient-specific therapies.
Esfahani et al. 2020, which provides a comprehensive review of cancer immunotherapy advancements, including CATI and CAR-M therapy.
Sequencing platforms


현재 Illumina는 NovaSeq 6000 모델이 주로 쓰인다. 이틀,48시간 만에 6테라를 생산할 수 있다. 이 데이터를 생산해도 우리가 가지고 있는 컴퓨터로는 어림도 없다. 전문적인 기기가 필요할 것이다.(워크스테이션이라던지...)


이건 중국산. 역시 아니나 다를까, Illumina를 그대로 모방했다. 이미테이션이라 하겠다. 그걸 인지했는지 가격은 Illumina보다 저렴하다. 가격으로 승부를 보겠다는 것?

Revio. 이 제품만 기억하자. 굉장히 길게 읽는다. 제일 긴 건 20kb까지 읽을 수 있다. 끊어지지 않고. 심지어 정확하다.
Illumina는 기껏해야 150bp인 것을 보라...
Illumina는, 이 게시글에서 앞서 설명했던 반응 reaction을 150번 정도 한다. 150번 밖에 못읽지만 필터링도 필요없을 정도로 매우 정확하다. 그냥 다른 작업 필요없이 바로 써도 될 정도이다. 근데 Revio 이 놈은 길게 읽는데 불구하고 버금가게 정확하기까지 하다.
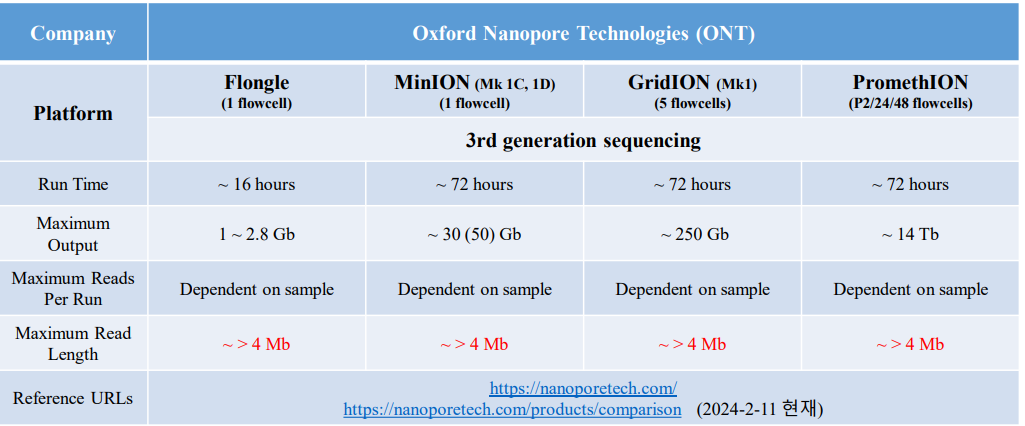
Nanopore 이 친구는 휴대폰보다도 작다. 그런데도 4mb까지 읽는다. 즉, DNA가 끊어지지만 않으면 다 읽을 수 있다. 하지만 에러가 많다는 것이 단점이다. 그래서 식물처럼 repeat sequence가 많은 경우 아니면 잘 쓰이지 않는다. 또는, 미생물의 경우 이를 사용하면 한 방에 다 읽을 수 있다. 그래서 이를 여러 cycle을 계속 돌리고 오버랩하면 정확해질 것이다.
위의 4가지 회사 외에는 이제 sequencing 회사가 없다.
'BIOLOGY > Bioinformatics' 카테고리의 다른 글
| 데이터 만들기 a.k.a 데이터 생산 (3) | 2024.10.19 |
|---|---|
| Sequencing에 점수를, Phred score (0) | 2024.10.18 |
| 저는 NGS를 하고 싶어요. (0) | 2024.10.18 |
| 바이오 빅데이터의 종류와 활용 (4) | 2024.10.13 |
| 생물학, 그리고 코딩을 곁들인 (1) | 2024.09.06 |
블로그의 정보
그럼에도 불구하고
Ungbae