![[팀 프로젝트] 데이터 모래에서 바늘 찾기](https://img1.daumcdn.net/thumb/R750x0/?scode=mtistory2&fname=https%3A%2F%2Fblog.kakaocdn.net%2Fdn%2FkX6cD%2FbtsM8b0qs5R%2Fy3KH6DkN2qpUAtukbcjAk0%2Fimg.png)

본 게시글은 학부 '생물정보학(Bioinformatics)' 강의 교수님의 지도 하에 진행된 팀 프로젝트입니다.
먼저 나는 태어나서 처음 접한 이 NCBI라는 사이트에 적혀있는 말들이 무슨 뜻인지 부터 알 필요가 있었다. 말도 안되는 양의 생물학 데이터들이 즐비한 이 곳에서 구글처럼 다루기 위해서라면.
Phase 2. 원하는 데이터 다운받기
본격적으로 우리가 정한 조사 주제를 하기 전에 NCBI를 둘러보고 어떻게 써먹어야 하는지를 알기위해 사용법을 익혀보았다.

잘 알려진 유글레나(Euglena)에 대해 검색을 해보자. 웬만하면 Long read가 좋다. Euglena pacbio라고 검색한 결과창이다.
결과창 오른쪽 중간을 보면 이 표를 볼 수 있다. BioProject의 public 란에 3개가 있다고 나와있다. 즉, 공개된 프로젝트가 3개가 있다고 보면 된다. 그래서 이 3에 걸려있는 링크를 타고 들어가보자.
링크를 타고 들어갔더니 The Euglena Genome Project 가 있는 것을 볼 수 있다. Human Genome Project 처럼 Euglena의 genome을 조립하기 위해서, 유전체 정보를 만들기 위해서 사람들이 데이터를 생산해 놓은 것이다. 그러면 우리는 이렇게 필요한 부분에서 검색해서 찾을 수 있게 되는 것.
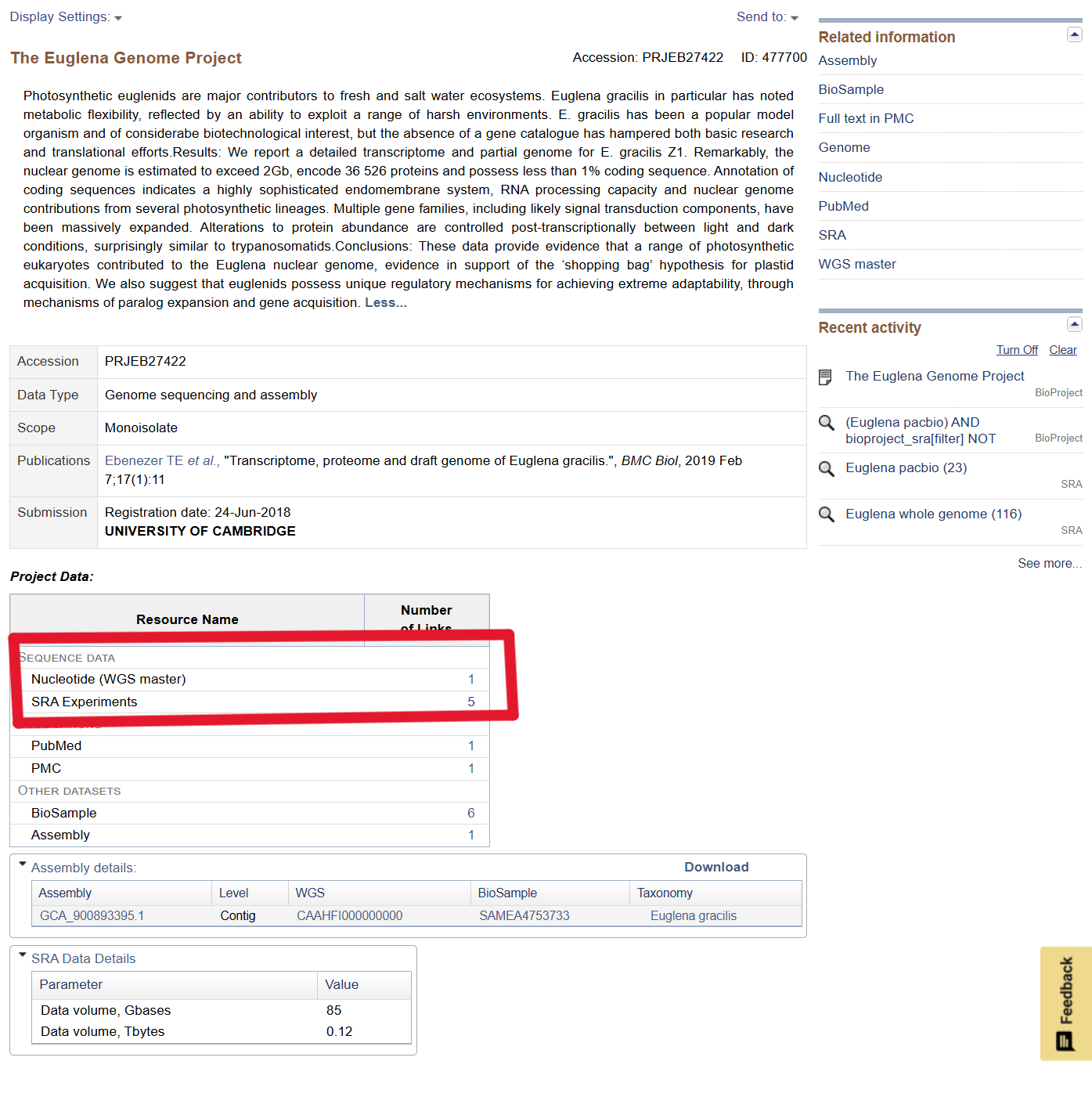
들어가봤더니 프로젝트에 대한 내용이 간략히(?) 설명되어 있다. 그 눈에 보이지 않는 생물에서도 2Gb나 되는 핵유전체를 가지고 있단다... 앞으로 무시하지 말자.
그리고 빨간 네모칸으로 표시한 곳 중에 SRA Experiments에 있는 5라는 number link를 들어가보자.
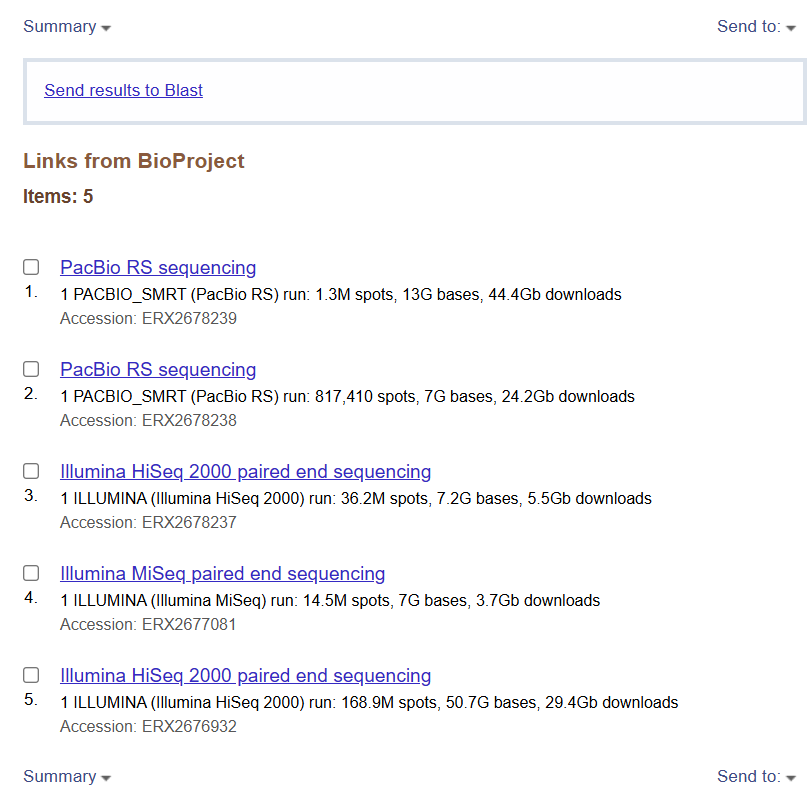
그 결과 이러한 목록을 찾을 수 있다.
첫 번째 PacBio RS sequencing을 볼 수 있고 그 밑에 대략적인 정보들이 검은 작은 글씨로 명시되어 있다.
PacBio라는 회사에서 만든 RS라는 장비로 생산한 데이터가 44.4G bases임을 알려주고 있다. 데이터가 44.4Gb이고 base로 보면 13Gb라는 것. 여기서 Giga는 10의 9승을 의미한다.
그런데 2번에도 똑같이 PacBio RS sequencing 데이터가 있다. 한 번 가지고는 부족했던 것이다. 두 번의 생산을 통해서 만들어냈음을 알 수 있다.
세 번째 Illumina HiSeq 2000 paired end sequencing을 보면 이제 무엇을 뜻하는지 보일 것이다.
Illumina의 HiSeq 2000이라는 장비를 사용하였고 제목을 보니 paired end라고 했기 때문에 양쪽으로 시퀀싱했다는 것을 알 수 있다. 다섯 번째 데이터에도 똑같은 제목이 있는 것으로 보아 이또한 한 번 더 생산했음을 알 수 있다.
생산량이 조금씩 다를 수 있다. 부족하면 더 생산할 수도 있고, 안 부족하면 이제 그만 생산해도 되는데 이게 같은 것들이 여러개 있다고 당황하지 말자.
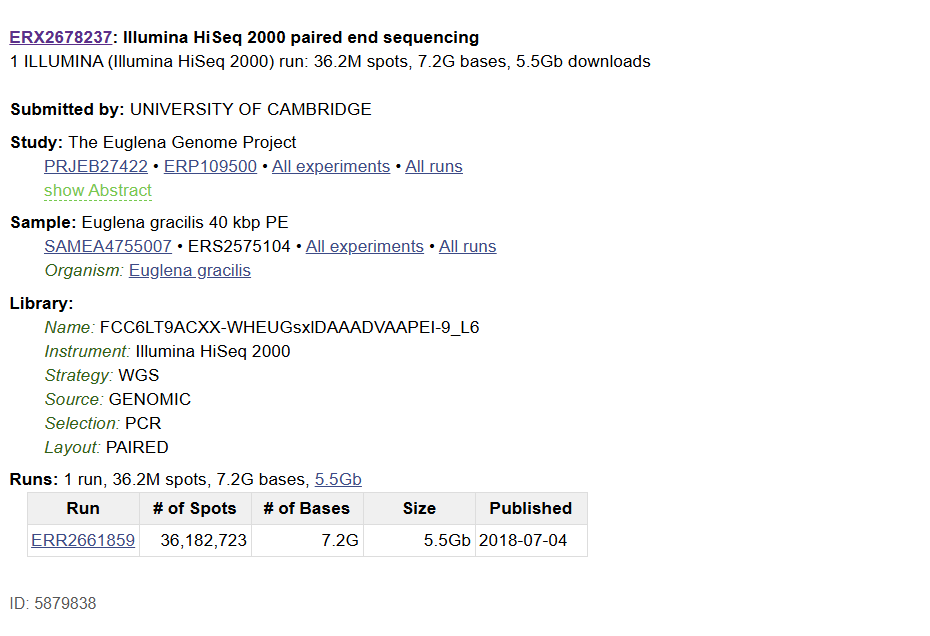
다섯 개 중 하나를 들어가 보았다. Library 카테고리에서 Strategy 란에 WGS라는 문구를 볼 수 있다. 이는 Whole Genome Sequence, DNA 전체를 다 읽었음을 의미한다. (꿈의 대학) 캠브리지, 영국에서 2018년 7월에 했다고도 명시되어 있다.

그 중 All runs를 클릭
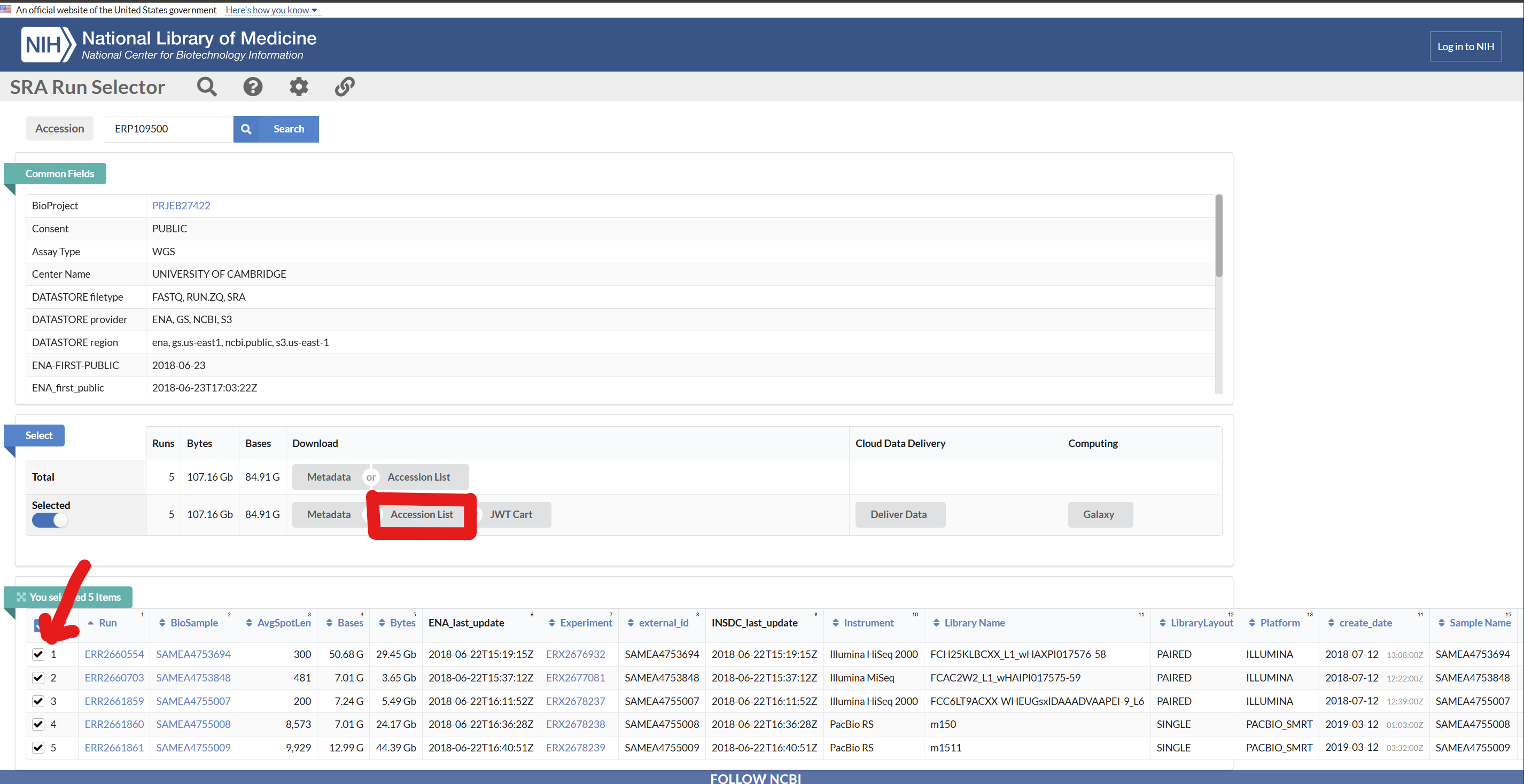
관련된 ERR 파일들, 즉 시퀀싱 데이터들을 한 번에 체크하고(빨간 화살표) 빨간색 상자가 가리키고 있는 Accession List를 누르게 되면 파일이 다운로드가 된다.
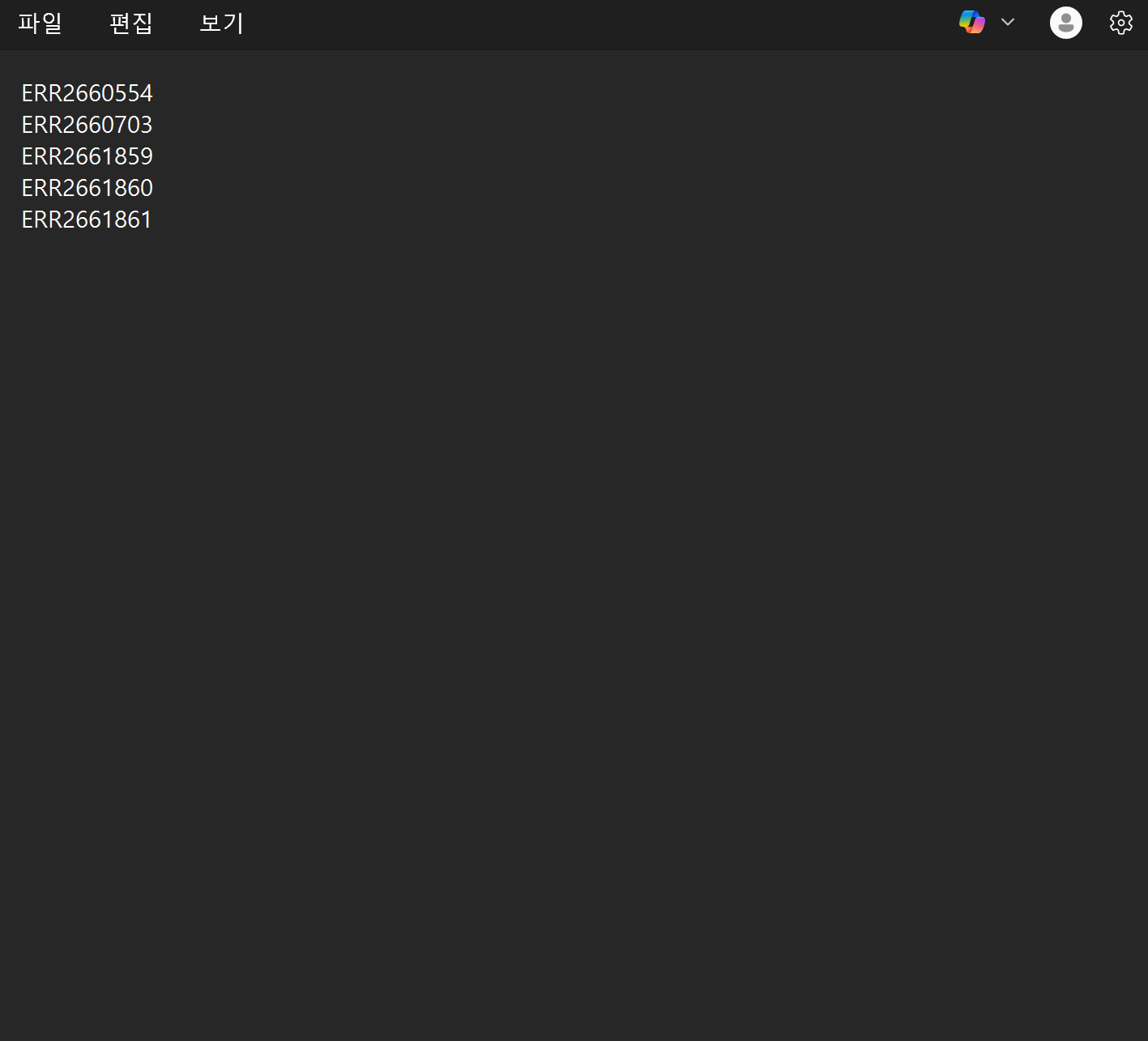
다운로드가 끝나면 이렇게 .txt 확장자 파일로 이렇게 ERR코드가 써있는 텍스트 파일이 다운로드 된 것을 확인할 수 있다.
우리는 이제 이 ERR 코드를 이용해서 리눅스를 통해 분석할 것이다.
<용어정리>
Specific gene = DEG = 특이반응유전자
SNP Calling : 유전변이를 찾아내는 것
DEG : Differentially Expressed Genes
" Specific gene을 찾는 가장 용이한 방법은 전사체 분석이다. 전사체를 통해 발현량을 볼 수 있기 때문이다. "
이제 준비는 끝났으니 본인의 리눅스 서버로 넘어가 파일을 다운로드 받아보자.
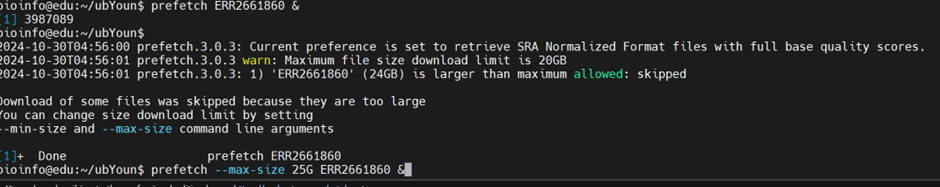
용량이 20G를 넘어갔다면 옵션 -max-size를 입력하여 큰 용량도 다운받을 수 있도록 하자.
<생물학 상식>
Satelite DNA는 microsatellite, minisatellite 등이 있다.
반복되는 염기서열의 수가 10개 이하일 경우 microsatellite.
e.g. AGCT가 4번 반복했다면? AGCT AGCT AGCT
-> 반복되는 서열은 맞으나 유전변이라 간주하지 않는다.
유전변이로 간주하려면?
e.g. 사람 A가 AGCT가 3번 반복했고, 사람 B는 AGCT가 5번 반복했다고 하자. A, B 사이에 같은 반복 서열이지만 다르다. 비교했을 때 다르기 때문에 이것이 개체 사이에 변이가 되는 것이다. 그렇다면, AGCT로 2번이 반복되었다면 반복되는 횟수가 다르기 때문에 유전 변이라고 한다. 여기서 반복되는 unit이 반복되는 수가 10번 미만.
하지만 300bp가 반복되고 이런 경우가 발현되면 이것을 통틀어서 satellite라고 한다. 사람마다 반복횟수가 다를 수 있다. 인종마다, 부모님 사이에서도 다를 수 있다.
다른 아버지와 어머니 사이에서도 각각 다른 반복된 서열을 받았다면?
A를 보았을 때 어머니가 AGCT가 네 번 반복한다고 하고, 아버지는 AGCT로 세 번 반복한다고 하자. A는 둘 다 가지고 있어야할 것이다. 이 영역을 14종류 조사한다. Satellite DNA는 수천만 군데에 존재한다. 여기서 다른 부분을 조사하는 것이다.
Retrotransposons
LTR, DNA 사이로 끼어 들어간다. Jumping DNA라고도 한다. Genome duplication 되는 과정 중에 하나. 누구는 삽입이 되었고 누구는 안되거나, 서로 다른 곳에 들어가는 경우 등에 의해 유전변이가 발생할 수 있는 것이다. 즉, Insertion과 Deletion. 합쳐서 In Del이라고 표현한다.
SNP
돌연변이로 인해서 Nucleotide가 랜덤하게 한 개 바뀌었다. 생식세포에 생겼는지, 체세포에 생겼는지 등의 여부를 알아내기 위해 유전체를 위해 우리는 유전체를 조립해야 한다.
서로 다르지 않으면 변이가 아니다. 변이는 달라야 한다. 개체 수준에서. 우리가 다른 것은 의미가 없다. 너와 내가 달라야 한다. 그 다름 때문에 겉으로 모습이 달라진다. 안경을 쓰고 안쓰는 등의 경우들이 변이에서 나타나는 것이다. 즉, 변이를 찾으면 안경을 쓰거나 안 쓴 그룹 간에 그 다름을 찾아내면 왜 안경을 쓰는 지에 대한 이유를 찾아낼 수 있다. 이를 고치면 안경을 쓰지 않게 되는 아이를 낳을 수 있을 것이다. 이것이 미래의학의 패러다임 중 하나일 것이다.
본 프로젝트 FASTA, FASTQ 파일 찾기용 검색 키워드
- 염증 반응 연구
- Homo Sapiens IL6 inflammation RNAseq
- Homo Sapiens TNF inflammation RNAseq
- 면역 세포 기반 데이터
- Homo Sapiens IL6 macrophage RNAseq
- Homo Sapiens TNF T cell RNAseq
- 질병 모델(감염 또는 면역 관련 질병 모델 데이터 찾기)
- Homo Sapiens IL6 COVID-19 RNAseq
- Homo Sapiens TNF sepsis RNAseq
'BIOLOGY > Bioinformatics' 카테고리의 다른 글
| [팀프로젝트] 프로젝트의 끝, 생어의 의지를 이어받다. (0) | 2025.04.09 |
|---|---|
| [팀프로젝트] 유전자를 디지털 월드로 (0) | 2025.04.08 |
| [팀프로젝트] 팀 프로젝트의 서막 (0) | 2024.12.01 |
| 데이터 수집부터 Alignment까지 (0) | 2024.10.22 |
| DNA 온라인 스토어 (4) | 2024.10.20 |
나의 성장 드라마
포스팅이 좋았다면 "좋아요❤️" 또는 "구독👍🏻" 해주세요!
![[팀프로젝트] 팀 프로젝트의 서막](https://img1.daumcdn.net/thumb/R750x0/?scode=mtistory2&fname=https%3A%2F%2Fblog.kakaocdn.net%2Fdn%2FW6Sks%2FbtsLaA3lpDG%2FX8MdXzMKVhpKjhOvZp4SX0%2Fimg.png)